Huntington's disease
Huntington’s disease affects around 5–10 people in 100,000 throughout developed countries. It is caused by the deterioration of various brain regions, including the cerebral cortex and basal ganglia. This results in uncontrollable body movements and stiffening of limbs. It can also cause emotional and cognitive impairments, including dementia and loss of memory, judgement and awareness.
Although there is an increasing number of people diagnosed with Huntington’s disease without any family history, it is a genetic condition that a child inherits from their parent. Only one parent needs to carry the gene mutation that causes the condition, and the children of affected parents have a 50/50 chance of inheriting the disease gene. If the gene mutation is inherited, they will develop Huntington’s disease.
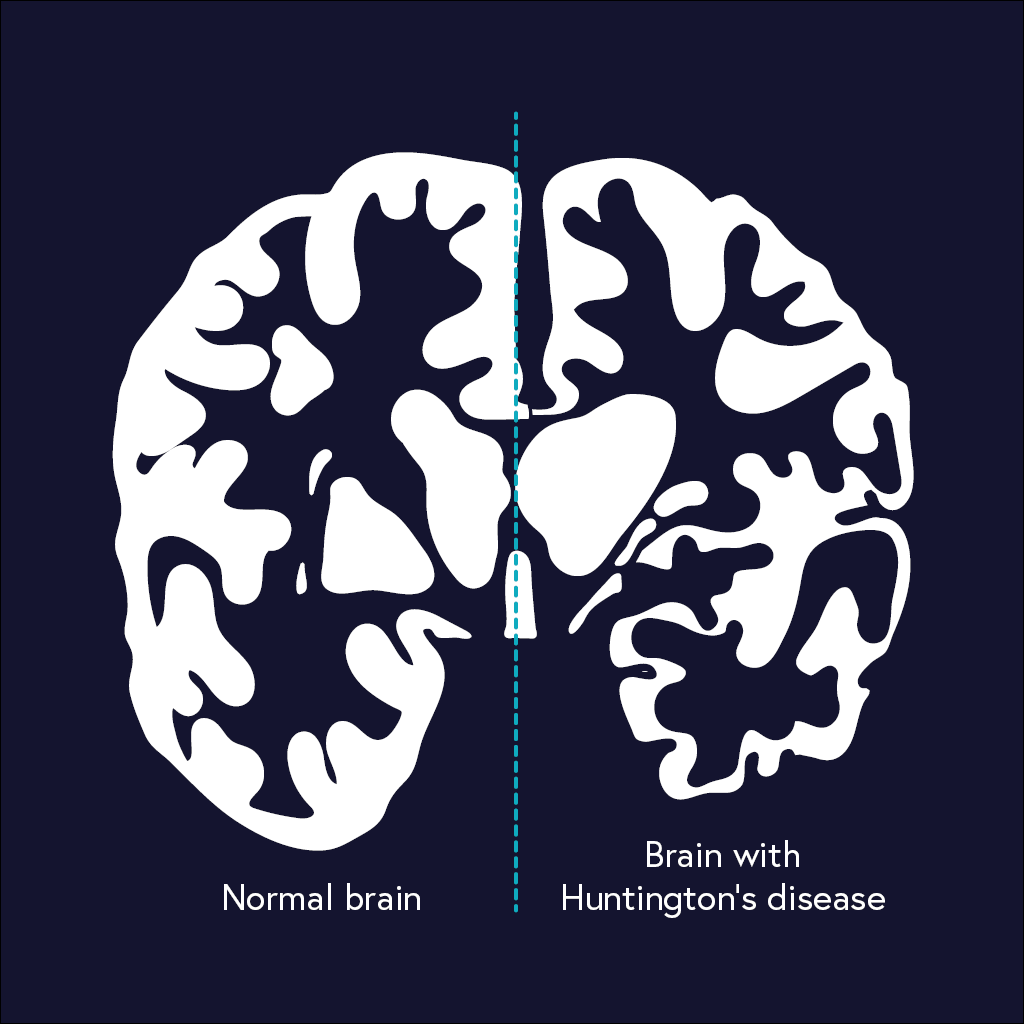
Huntington’s disease usually manifests when people are in their 40s or 50s. The earlier the symptoms appear, the faster the disease seems to progress. Huntington’s disease is progressive, meaning it worsens over time. The expected lifespan for people with the condition once symptoms appear is 10–25 years.
There are treatments that help control some symptoms, but there is no disease-modifying treatment or cure for Huntington’s disease.
The Huntington’s disease gene includes a ‘genetic stutter’ that causes a ‘tandem repeat disorder’, where a sequence of three DNA nucleotides—cytosine, adenine and guanine, or CAG—are repeated too many times. When this DNA repeat is translated into protein it makes an abnormally long tract of the amino acid glutamine, repeated within the huntingtin protein. This polyglutamine tract is toxic to specific types of neurons and also other cells in the brain and body.
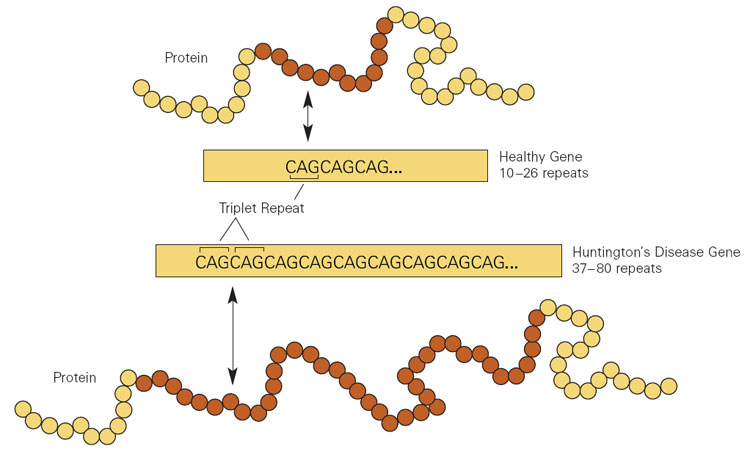
Huntington’s disease is one of over 30 tandem repeat disorders which include other diseases as diverse as spinocerebellar ataxias, Kennedy’s disease, fragile X syndrome, and a subset of motor neuron disease and frontotemporal dementia.
The expanded polyglutamine tract in the huntingtin protein can disrupt the expression of many other genes and impair the transport of proteins and other molecules around cells. It can also affect the function of synapses, which are crucial in brain circuits mediating various aspects of cognition, emotion and movement.
Huntington’s disease research currently involves a wide range of disease models, as well as clinical studies. Much of the research focuses on how specific molecules and cells are disrupted, and how they give rise to the movement disorder, psychiatric symptoms and cognitive deficits, which culminate in dementia.

The research field is rapidly moving to establish sophisticated models of disease mechanisms, develop new treatments and establish clinical trials. An active area of research is the development of pluripotent stem cell lines, where cells from a Huntington’s disease patient are ‘forced’ to revert back to an early stage of cell development, then made to develop into brain cells that have the characteristics of the disease. This enables researchers to study the disease progression and investigate new options for treatments. There are other exciting developments on the cards with clinical trials underway for a drug that can potentially silence the huntingtin gene, preventing the disease from manifesting.
Other options for treatment involving neurotechnology applications are also being explored. These could be used to prevent or ameliorate the movement disorder, and psychiatric or cognitive problems.